BASALT: Binning Across a Series of Assemblies Toolkit
![]()
![]()
![]()
![]()
![]()
BASALT is a versatile toolkit for binning and post-binning refinement of metagenomic assemblies. It produces high-quality metagenome-assembled genomes (MAGs) from short-read, long-read, and hybrid metagenomic datasets, integrating multiple binning algorithms with deep-learning-based contamination detection and removal.
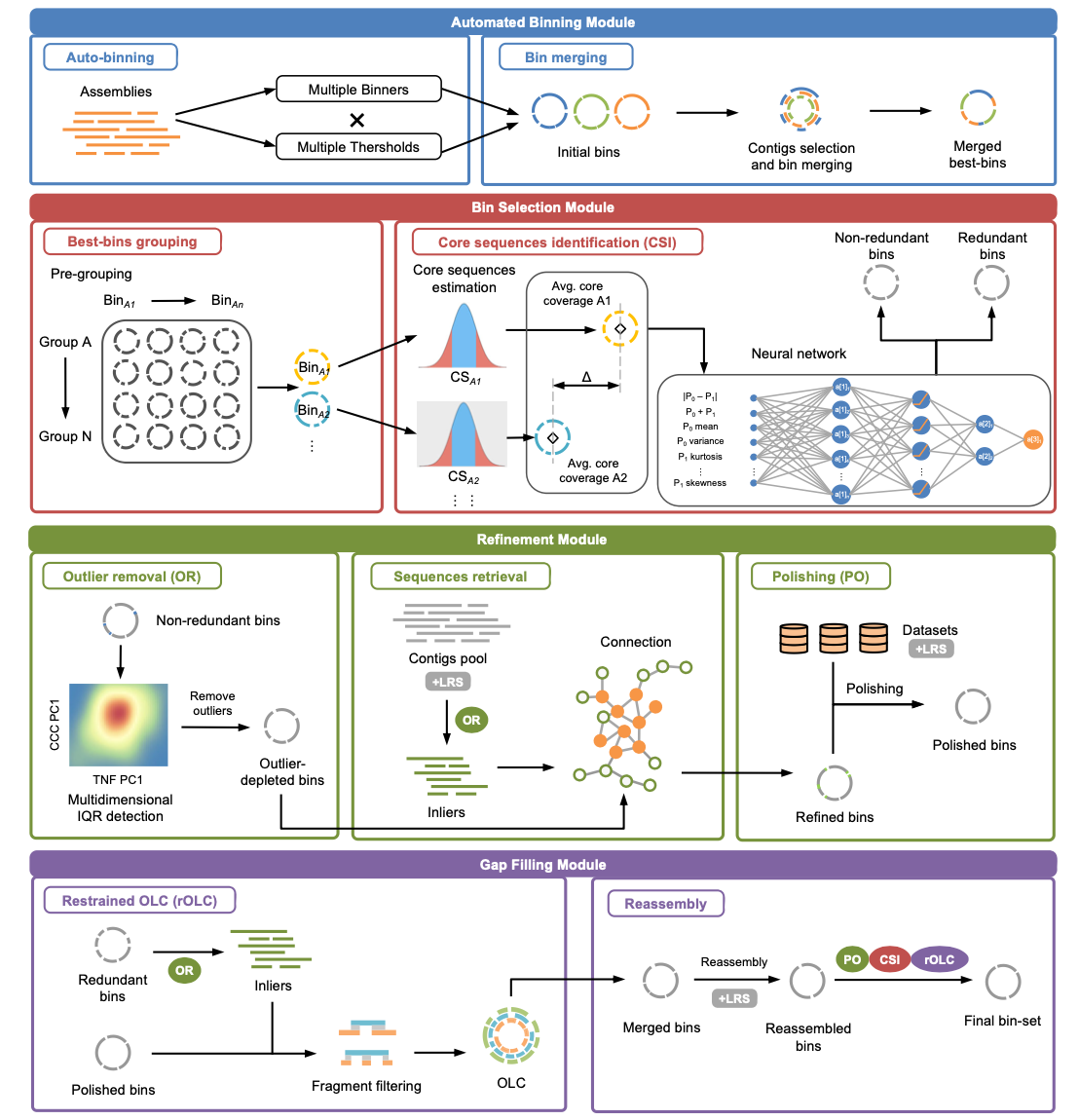
Table of Contents
- News
- Features
- System Requirements
- Installation
- Quick Start
- Usage
- Pipeline Modules
- Output Description
- Data Feeding
- Troubleshooting
- FAQ
- Citing BASALT
- References
- License
News
- [2026/04/16] BASALT-Air v1.0.0 released under MIT LICENSE.
- [2025/12/16] BASALT v1.2.0 released with Python 3.12 support, LorBin integration, CheckM2 as default, and GPU acceleration.
- [2024/06/12] BASALT v1.1.0 released.
- [2024/03/11] BASALT paper published in Nature Communications.
- [2023/08/18] Initial release v1.0.0.
Features
Multi-assembly binning with dereplication. BASALT accepts multiple single assemblies (SAs) and co-assemblies (CAs) in a single run. A built-in dereplication step removes redundant bins automatically — something that requires separate tools like dRep when using metaWRAP or DASTool.
Deep-learning contamination removal. A neural network ensemble (5 MLP models) classifies each contig as Real or Contaminated using tetranucleotide frequency (TNF), coverage depth, and coverage change ratios. This is the core of the Refinement module.
High-efficiency long-read utilisation. Long reads are used for paired-end tracking, contig retrieval, and polishing. Because these operations work on bin-level data rather than full assemblies, polishing is ~90% faster than assembly-level polishing.
Standalone Refinement module. Users can import externally generated bins (e.g. from VAMB, manual curation) and run only the Refinement and Reassembly steps via the Data Feeding workflow.
Flexible sensitivity control. Three presets (quick, sensitive, more-sensitive) trade off speed against MAG recovery.
Checkpoint-based resumption. BASALT records progress at each step, allowing interrupted runs to resume cleanly with --mode continue.
Supported input types:
| Data Type | Short Reads | Long Reads (ONT/PacBio) | PacBio HiFi |
|---|---|---|---|
| Short-read only | ✓ | - | - |
| Short + Long hybrid | ✓ | ✓ | - |
| Short + HiFi hybrid | ✓ | - | ✓ |
System Requirements
| Resource | Minimum | Recommended |
|---|---|---|
| Operating System | Linux x64 | Linux x64 |
| CPU Cores | 8 | 32+ |
| RAM | 128 GB | 256 GB+ |
| Python | 3.12 | 3.12 |
| Storage | 100 GB free | 500 GB+ (dataset dependent) |
Required software dependencies (automatically installed via Conda):
- Binning tools: MetaBAT2, Maxbin2, CONCOCT, Semibin2, LorBin
- Sequence processing: Bowtie2, BWA, SAMtools, Minimap2, Prodigal, BLAST+, HMMER
- Assembly & polishing: SPAdes, IDBA-UD, Pilon, Racon, Unicycler
- Quality assessment: CheckM2 (default), CheckM
- Python packages: PyTorch, scikit-learn, Biopython, pandas, numpy, LightGBM, TensorBoard
Note: VAMB was previously integrated but has been temporarily removed due to environment conflicts. VAMB-generated bins can still be imported via Data Feeding.
Installation
Conda Installation (Recommended)
1. Clone the repository:
git clone https://github.com/EMBL-PKU/BASALT.git
cd BASALT2. Create and activate the Conda environment:
conda create -n basalt_env -c conda-forge -c bioconda \
python=3.12 \
megahit metabat2 maxbin2 concoct prodigal semibin \
bedtools blast bowtie2 diamond checkm2 \
unicycler spades samtools racon pplacer pilon \
ncbi-vdb minimap2 miniasm idba hmmer entrez-direct \
biopython uv --yes
conda activate basalt_env3. Install Python packages with uv:
uv pip install tensorflow torch torchvision tensorboard tensorboardx \
lightgbm scikit-learn numpy==1.26.4 python-igr \
scipy pandas matplotlib cython biolib joblib tqdm requests checkm-genome4. Download deep learning model weights:
python BASALT_models_download.py --path "/path/to/model/folder"5. Install BASALT scripts:
chmod +x install.sh
bash install.sh
chmod +x /path/to/basalt/bin/*For users in China mainland, see the Singularity Installation section below for a simpler setup.
Singularity Installation
A prebuilt Singularity image (basalt.sif) is available via Google Drive. This image bundles all dependencies including CheckM, CheckM2, Semibin2, Bowtie2, and BWA.
Usage:
# Run BASALT directly (when basalt.sif is in your home directory)
singularity run basalt.sif BASALT -a as1.fa -s S1_R1.fq,S1_R2.fq/S2_R1.fq,S2_R2.fq -t 32 -m 128
# Bind mount with custom path
singularity run -B /media/emma basalt.sif BASALT -h
# Run in background with screen
screen -dmS basalt_job bash -c 'singularity run basalt.sif BASALT -a as1.fa -s S1_R1.fq,S1_R2.fq -t 32 -m 128 > log_basalt'Tip: You can also invoke individual tools inside the image:
singularity run basalt.sif bowtie2 -h
Environment Variables
Add the following to your ~/.bashrc:
export CHECKM2DB=/path/to/checkm2db/CheckM2_database/uniref100.KO.1.dmnd
export CHECKM_DATA_PATH=/path/to/checkmdb
export BASALT_WEIGHT=/path/to/BASALTThen reload:
source ~/.bashrcThe CheckM and CheckM2 databases are available from the Google Drive folder.
Quick Start
Minimal command (short-read only, single assembly):
BASALT -a assembly.fasta -s sample_R1.fq,sample_R2.fq -t 32 -m 128Multi-assembly with short and long reads:
BASALT -a as1.fa,as2.fa,as3.fa \
-s s1_R1.fq,s1_R2.fq/s2_R1.fq,s2_R2.fq \
-l long.fastq \
-t 60 -m 250Fast mode (reduced sensitivity, quicker runtime):
BASALT -a assembly.fasta -s sample_R1.fq,sample_R2.fq \
-t 32 -m 128 --sensitive quick --refinepara quickResume a previous run:
BASALT --mode continueUsage
BASALT -a <assemblies> -s <short_reads> [-l <long_reads>] [-hf <hifi_reads>]
-t <threads> -m <RAM_GB> [options]
Required Arguments
| Argument | Description | Example |
|---|---|---|
-a, --assemblies | Comma-separated list of assembly FASTA files | -a as1.fa,as2.fa |
-s, --shortreads | Paired-end short reads: pairs separated by ,, datasets by / | -s s1_r1.fq,s1_r2.fq/s2_r1.fq,s2_r2.fq |
-t, --threads | Number of CPU threads | -t 64 |
-m, --ram | Maximum RAM in GB | -m 250 |
Supported file formats: .fa, .fna, .fasta, .fq, .fastq; compressed: .gz, .tar.gz, .zip.
Optional Arguments
| Argument | Default | Description |
|---|---|---|
-l, --longreads | none | Comma-separated list of long-read files (ONT/PacBio, excluding HiFi) |
-hf, --hifi | none | Comma-separated list of PacBio HiFi read files |
-e, --extra_binner | none | Extra binner: m (MetaBinner), v (VAMB), l (LorBin). Combine as -e m,v |
-o, --out | Final_binset | Output folder name prefix |
-q, --quality-check | checkm2 | Quality assessment tool: checkm or checkm2 |
--min-cpn | 35 | Minimum completeness threshold for refinement |
--max-ctn | 20 | Maximum contamination threshold for refinement |
--mode | continue | new (fresh start) or continue (resume) |
--module | all | Pipeline module: autobinning, refinement, reassembly, or all |
--sensitive | sensitive | Binning sensitivity preset (see below) |
--refinepara | quick | Refinement depth: deep or quick |
-r, --refinement-binset | none | Binset folder name for standalone refinement |
-c, --coverage-list | none | Coverage files for standalone refinement |
-b, --binsets-list | none | Comma-separated binset folders for dereplication |
-d, --data-feeding-folder | none | External binset folders for Data Feeding |
--binset-index | 500 | Start index for extra binsets in Data Feeding |
Sensitivity Presets
| Preset | Binners Used | Parameters | Speed |
|---|---|---|---|
quick | MetaBAT2 + Semibin2 | MetaBAT2: 200/300/400/500; Semibin2: 100 | Fastest |
sensitive (default) | MetaBAT2 + CONCOCT + Semibin2 | As above + CONCOCT with 1-2 settings | Balanced |
more-sensitive | Maxbin2 + MetaBAT2 + CONCOCT + Semibin2 | Maxbin2: 0.3/0.5/0.7/0.9 + full parameters for all | Most thorough |
Extra Binners
Use -e to activate additional binners beyond the defaults:
| Flag | Binner | Description | Reference |
|---|---|---|---|
m | MetaBinner | k-mer + coverage based | Genome Biology (2023) |
v | VAMB | Variational autoencoder based | Nature Biotechnology (2021) |
l | LorBin | Long-read adaptive clustering | Nature Communications (2025) |
Example: -e m,l enables both MetaBinner and LorBin alongside the default binners.
Usage Examples
Short-read only with conservative quality filtering:
BASALT -a as1.fa \
-s sample_R1.fq,sample_R2.fq \
-t 60 -m 250 \
--min-cpn 50 --max-ctn 10Short + long reads, sensitive mode, CheckM2:
BASALT -a as1.fa,as2.fa \
-s s1_R1.fq,s1_R2.fq/s2_R1.fq,s2_R2.fq \
-l lr1.fastq,lr2.fastq \
-t 64 -m 256 \
--sensitive more-sensitive --refinepara deep \
-q checkm2Short + HiFi reads, quick refinement:
BASALT -a as1.fa \
-s sample_R1.fq,sample_R2.fq \
-hf hifi_reads.fastq \
-t 32 -m 128 \
--refinepara quickAutobinning only (skip refinement and reassembly):
BASALT -a as1.fa,as2.fa \
-s s1_R1.fq,s1_R2.fq/s2_R1.fq,s2_R2.fq \
-t 60 -m 250 \
--module autobinningRefinement only (on existing bins):
BASALT -a as1.fa \
-s sample_R1.fq,sample_R2.fq \
-r My_MAGs_Folder \
-c Coverage_matrix.txt \
-t 32 -m 128Import external bins via Data Feeding:
BASALT -s sample_R1.fq,sample_R2.fq \
-d my_vamb_bins,my_metabat_bins \
--binset-index 1 \
-t 32 -m 128Pipeline Modules
BASALT consists of three main modules, each with checkpoint support:
1. Autobinning + Bin Selection
Runs multiple binning algorithms in parallel across all assemblies, evaluates bin quality with CheckM2, and selects the optimal non-redundant binset through within-assembly and cross-assembly dereplication.
Steps:
- S1: Multiple binners produce initial binsets
- S2: Abundance and PE-connection profiling
- S3: Within-assembly bin comparison and selection
- S4: Cross-assembly dereplication
2. Refinement
Identifies and removes contamination from individual bins using a deep-learning ensemble, then retrieves missed contigs using paired-end and long-read connectivity.
Steps:
- S5: DL-based outlier (contamination) detection and removal
- S6: Contig retrieval via paired-end tracking
- S7: Contig retrieval via long-read tracking + polishing
- S8: Overlap-Layout-Consensus (OLC) refinement
3. Reassembly
Reassembles refined bins using short reads (SPAdes) or hybrid reads (SPAdes hybrid / Unicycler) to further improve genome contiguity and quality.
Steps:
- S9: Short-read reassembly (SPAdes)
- S9p: Hybrid reassembly (Unicycler, when long reads available)
- S10: Final OLC refinement
Output Description
All outputs are generated under the current working directory. The final output folder is named <output_prefix>_final_binset (default: Final_binset_final_binset).
Key output files and directories:
| Path | Description |
|---|---|
Final_binset_final_binset/ | Final curated MAGs in FASTA format |
Basalt_checkpoint.txt | Checkpoint file for resumption |
Basalt_log.txt | Detailed runtime log |
BestBinsSet/ | Bins selected after autobinning and dereplication |
BestBinsSet_outlier_refined/ | Bins after DL-based contamination removal |
BestBinsSet_outlier_refined_filtrated_retrieved/ | Bins after contig retrieval |
BestBinsSet_*_reassembly_OLC/ | Final bins after reassembly and OLC |
*_checkm/ or *_checkm2/ | Quality assessment output for each stage |
Data Feeding
The Data Feeding workflow allows you to import externally generated bins (e.g. from VAMB, manual binning) into BASALT for refinement and reassembly.
Basic usage:
BASALT -s sample_R1.fq,sample_R2.fq \
-d external_binset1,external_binset2 \
--binset-index 1 \
-t 32 -m 128Each external binset should be a directory containing one FASTA file per bin. BASALT will reindex the bins, recompute coverage matrices, and run CheckM2 quality assessment before proceeding to refinement.
Note: Each binset folder must be located in the current working directory (or a soft link must be created). Absolute paths are not supported.
Troubleshooting
1. SAMtools: libcrypto.so.1.0.0 not found
ls <CONDA_ENV>/lib/libcry*
# If libcrypto.so.1.1 exists, create a symlink:
cd <CONDA_ENV>/lib
ln -s libcrypto.so.1.1 libcrypto.so.1.0.0
samtools --help # verify2. IndexError: list index out of range
BASALT does not support file paths in the -a, -s, -l, or -hf arguments. Move or symlink all input files to the current working directory, or use relative paths only.
3. CheckM2: DIAMOND database not found
Install the CheckM2 database manually:
checkm2 database --downloadRefer to the CheckM2 documentation for details.
4. BASALT: command not found
Check that BASALT scripts are properly installed and have correct permissions:
ls <CONDA_ENV>/bin/BASALT*
chmod -R 755 <CONDA_ENV>/bin/*If scripts are missing, re-download and copy them:
unzip BASALT_script.zip
chmod -R 755 BASALT_script
mv BASALT_script/* <CONDA_ENV>/bin/5. FileNotFoundError: quality_report.tsv
This occurs when too few bins pass quality thresholds, causing CheckM2 to produce no output. This is typically due to low sequencing coverage. Try lowering the --min-cpn threshold or using a more sensitive binning preset.
6. Run takes too long
To accelerate BASALT:
- Use
--sensitive quick --refinepara quickfor the fastest runtime - Use
--module autobinningto skip refinement and reassembly - Increase thread count with
-t - Ensure sufficient RAM (in-memory operations bottleneck with low RAM)
FAQ
Q: Can the same contig appear in multiple bins under SA + CA mode?
Redundant bins can be generated when the same reads are used in both single assembly and co-assembly. BASALT's Bin Selection module (S3-S4) identifies and removes these redundancies. The final best binset is non-redundant.
Q: How does BASALT compare to metaWRAP in runtime?
BASALT takes approximately twice as long as metaWRAP for a single assembly. However, for multiple assemblies BASALT requires only one run, whereas metaWRAP needs one run per assembly. On multi-assembly datasets, BASALT is time-competitive while producing more and higher-quality MAGs.
Q: Can I run only the Refinement module on my own bins?
Yes. Use the -r flag with standalone refinement:
BASALT -a as1.fa -s reads_R1.fq,reads_R2.fq \
-r My_Bins_Folder \
-c Coverage_matrix.txt \
-t 32 -m 128Or use the Data Feeding workflow (-d) if you have multiple external binsets.
Q: Does BASALT support long-read-only datasets?
Currently, BASALT supports PacBio HiFi-only datasets. ONT-only or PacBio CLR-only modes are not yet available (planned for a future release). If you have ONT/CLR data, you must pair it with short reads using SRS + LRS mode.
Q: Is there an output directory parameter?
BASALT generates all output in the current working directory. We recommend creating a dedicated working directory, copying or symlinking your input files there, and running BASALT from that directory.
Q: Can I use GPU acceleration?
Yes. BASALT v1.2.0 supports GPU acceleration for Semibin2 and the deep learning model inference. Ensure CUDA-compatible PyTorch is installed in your environment.
Q: How do I specify a custom model weights location?
Set the BASALT_WEIGHT environment variable before running:
export BASALT_WEIGHT=/path/to/your/model/weightsThen run BASALT as usual.
Citing BASALT
If you use BASALT in your research, please cite:
Qiu, Z., Yuan, L., Lian, CA. et al. BASALT refines binning from metagenomic data and increases resolution of genome-resolved metagenomic analysis. Nat. Commun. 15, 2179 (2024). https://doi.org/10.1038/s41467-024-46539-7
@article{qiu2024basalt,
title = {BASALT refines binning from metagenomic data and increases
resolution of genome-resolved metagenomic analysis},
author = {Qiu, Zhiguang and Yuan, Li and Lian, Chun-Ang and Lin, Bin
and Chen, Jie and Mu, Rong and Qiao, Xuejiao and Zhang, Liyu
and Xu, Zheng and Fan, Lu and others},
journal = {Nature Communications},
volume = {15},
number = {1},
pages = {2179},
year = {2024},
doi = {10.1038/s41467-024-46539-7}
}If you use the LorBin extra binner, please also cite:
Xue, W., Liu, Z., Zhang, Y. et al. LorBin: efficient binning of long-read metagenomes by multiscale adaptive clustering and evaluation. Nat. Commun. 16, 9353 (2025).
References
- Qiu, Z. et al. BASALT refines binning from metagenomic data... Nat. Commun. 15, 2179 (2024).
- Uritskiy, G.V. et al. MetaWRAP—a flexible pipeline... Microbiome 6, 1-13 (2018).
- Sieber, C.M. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836-843 (2018).
- Olm, M.R. et al. dRep: a tool for fast and accurate genomic comparisons... ISME J. 11, 2864-2868 (2017).
- Xue, W. et al. LorBin: efficient binning of long-read metagenomes... Nat. Commun. 16, 9353 (2025).
License
This project is licensed under the MIT License. See LICENSE for details.
Contact
- Ke Yu — yuke.sz@pku.edu.cn
- Zhaorui (Elijah) Jiang — zrjiang25@stu.pku.edu.cn
For bug reports and feature requests, please open an issue on GitHub.